LPS와 알츠하이머병: 내독소 가설을 8편의 근거로 읽는 도슨트
발행: 2026-02-26 · 최종 업데이트: 2026-02-26
LPS와 알츠하이머병의 연결을 BBB, 아밀로이드 대사, 신경염증, 인간 코호트 근거까지 8편의 핵심 문헌으로 통합 정리합니다.
이 글의 질문
알츠하이머병(AD)은 전통적으로 아밀로이드와 타우 중심으로 설명되어 왔습니다. 그러나 최근에는 장내 또는 전신에서 유래한 저강도 내독소 신호, 특히 LPS가 신경염증과 병리 진행을 밀어 올릴 수 있다는 관점이 빠르게 확장되고 있습니다. 이 글은 “LPS가 AD 병리의 원인인가, 동반자인가”라는 질문을 8편의 핵심 논문으로 정리하는 docent 글입니다.
왜 지금 AD에서 LPS를 다시 보나
AD 연구는 오랫동안 뇌 안에서 일어나는 단백질 병리, 특히 Aβ 축적과 타우 인산화에 초점을 맞춰 왔습니다. 이 틀은 여전히 유효하지만, 임상 현장에서 관찰되는 이질성을 모두 설명하지는 못합니다. 같은 병리 부담을 가져도 진행 속도와 임상 표현형이 다르고, 전신 염증 상태에 따라 악화 속도가 달라지는 현상이 반복되기 때문입니다.
이 지점에서 LPS 가설은 “기존 이론의 대체재”라기보다 “빈칸을 메우는 연결축”으로 등장합니다. 즉, 장·폐·구강 등 말초의 미생물 유래 신호가 BBB, 미세아교세포, Aβ 처리 경로에 동시에 영향을 주어 뇌 병리를 가속할 수 있다는 모델입니다. 이 모델의 강점은 단일 분자 인과를 강요하지 않고, AD를 전신-뇌 네트워크 질환으로 읽게 만든다는 데 있습니다.
1) 기전 축: BBB와 Aβ 대사부터 흔들린다
Jaeger(2009)와 Erickson(2012) 계열 연구는 LPS가 혈뇌장벽(BBB) 기능과 아밀로이드 베타(Aβ) 수송을 교란할 수 있음을 보여줍니다. 핵심은 뇌 안에서 Aβ가 더 많이 만들어지는가만이 아니라, 이미 생성된 Aβ가 뇌 밖으로 제대로 빠져나가지 못하는 상황이 동시에 만들어질 수 있다는 점입니다. 즉, LPS는 “생성 증가 + 배출 저하”라는 이중 축으로 AD 병리의 임계치를 낮출 가능성이 있습니다.
2) 동물 근거: LPS는 인지저하와 병리 변화를 재현한다
Lee(2008), Erickson(2012), Hunter(2018) 등 동물실험에서 반복 LPS 노출은 인지기능 저하, Aβ 증가, 일부 모델에서 p-tau 증가까지 동반했습니다. 여기서 중요한 해석은 LPS가 AD를 단독으로 “완성”한다는 뜻이 아니라, 이미 취약한 뇌 환경에서 염증성 가속기 역할을 할 수 있다는 점입니다. 다시 말해 LPS는 점화 스위치라기보다 증폭기(amplifier)로 읽는 것이 현재 근거와 더 잘 맞습니다.
3) 인간 조직 근거: AD 뇌에서 LPS 관련 신호가 관찰된다
Zhan(2016) 연구는 AD 사후 뇌 조직에서 LPS 및 E. coli 관련 분자 신호가 증가하고, 이 신호가 아밀로이드 베타와 공간적으로 연관된다는 결과를 제시했습니다. 이 데이터의 강점은 “사람 뇌에서의 관찰”이라는 점이고, 약점은 관찰연구 특성상 인과 방향을 확정할 수 없다는 점입니다. 그럼에도 동물·기전 데이터와 결합하면, LPS가 AD 병리 환경에 실제로 개입할 여지가 있다는 신호로 해석할 수 있습니다.
4) 코호트 근거: 말초 내독소 지표와 장기 위험
André(2019)는 말초 내독소 관련 지표(LBP, sCD14)가 향후 AD 위험과 연결될 가능성을 제시했습니다. 이 결과는 “뇌 안에서만 보는 AD”에서 “전신 면역-대사 상태를 함께 보는 AD”로 관점을 넓혀 줍니다. 다만 이 역시 위험 연관성 데이터이므로, 임상적으로는 단일 바이오마커 확정이 아니라 위험층화 보조지표로 다루는 것이 안전합니다.
5) 통합 모델: 원인 단일론보다 네트워크 모델이 맞다
Brown & Heneka(2024) 리뷰가 주는 핵심은 단순합니다. LPS를 AD의 유일 원인으로 보기는 어렵지만, 미세아교세포 활성, BBB 기능 저하, Aβ 처리 실패, 전신 염증 신호를 연결하는 상위 네트워크 입력으로는 충분히 설득력이 있다는 점입니다. 즉, LPS 가설의 실제 가치는 “대체 이론”이 아니라 “연결 이론”에 있습니다.
논문들이 서로 충돌하는 지점은 어디인가
같은 “LPS-AD 연관”을 다루더라도 논문마다 질문이 다릅니다. 어떤 연구는 BBB를 보고, 어떤 연구는 Aβ 수송을 보고, 어떤 연구는 행동 변화나 코호트 위험을 봅니다. 그래서 한 논문의 결과를 다른 논문의 결론처럼 읽으면 과잉 해석이 생깁니다.
핵심 충돌은 크게 세 가지입니다. 첫째, 시간축입니다. 급성 LPS 노출 모델의 변화가 만성 신경퇴행을 얼마나 설명하는지 분리해야 합니다. 둘째, 공간축입니다. 말초 혈중 지표 상승이 실제 뇌실질 신호 증가를 얼마나 반영하는지 아직 정밀한 매핑이 부족합니다. 셋째, 인과축입니다. LPS가 병리의 시작점인지, 이미 진행된 병리에서 증폭기인지 구분이 어렵습니다. docent 글에서 중요한 점은 이 충돌을 지우는 것이 아니라, 독자가 “어디까지가 데이터이고 어디부터가 해석인지”를 구분해 읽게 만드는 것입니다.

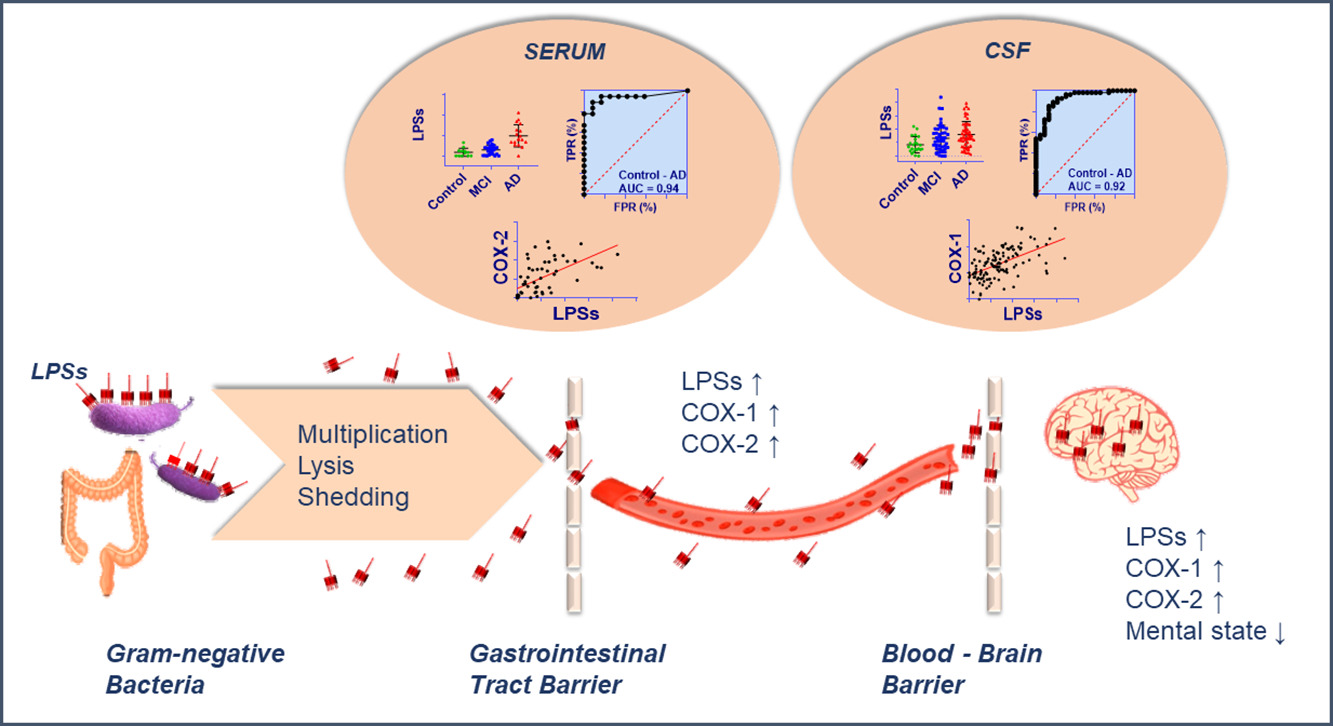
무엇이 확정되었고 무엇이 남았는가
현재 비교적 견고한 부분은 세 가지입니다. 첫째, LPS 노출이 BBB·신경염증·Aβ 대사에 영향을 줄 수 있다는 기전 축이 존재합니다. 둘째, 동물모델에서 LPS 노출은 인지 및 병리 악화와 일관된 방향성을 보입니다. 셋째, 인간 조직·코호트에서 LPS 관련 신호가 AD와 연관됩니다.
반대로 미확정 영역도 분명합니다. LPS 신호가 AD의 “시작 원인”인지, 병리 진행 과정에서 커지는 “증폭 요인”인지는 아직 확정되지 않았습니다. 또한 환자군 내 이질성, 감염원 다양성, 식이·대사·혈관 요인의 교란을 정교하게 분리하는 작업이 더 필요합니다.
실무적으로 이 문헌군을 어떻게 읽을까
이 주제를 실제 글쓰기나 연구 기획에 적용할 때는 세 가지 원칙이 유용합니다. 첫째, “LPS가 AD를 유발한다”가 아니라 “LPS가 AD 병리 네트워크를 증폭할 수 있다”로 표현 강도를 통제합니다. 둘째, 동물·사후조직·코호트 근거를 한 문단에서 섞지 말고 층위별로 분리해 제시합니다. 셋째, 바이오마커 해석에서는 단일 지표 확정보다 복합 위험도(염증지표, 혈관지표, 대사지표) 프레임으로 연결합니다.
이렇게 읽으면 과장 없이도 메시지 밀도를 높일 수 있고, 이후 정신건강·우울증·대사질환으로 이어지는 LPS-saga의 확장성도 유지할 수 있습니다.
LPS-saga에서의 위치
이 주제는 LPS-saga를 감염성 급성염증(패혈증) 서사에서 만성 신경퇴행 서사로 확장하는 연결 고리입니다. 같은 LPS라도 맥락이 달라지면 임상 표현형이 달라진다는 점, 즉 “고강도 급성 독성”과 “저강도 만성 증폭”이 공존할 수 있다는 점을 가장 선명하게 보여줍니다.
결론
알츠하이머병에서 LPS 가설은 과장도 과소평가도 피해야 합니다. 현재 근거 수준에서 가장 안전한 결론은, LPS가 AD를 단독으로 설명하지는 못하지만 병리 네트워크를 가속할 수 있는 유력한 염증성 입력이라는 것입니다. 그래서 이 주제는 단일 논문보다, 기전-동물-인간 근거를 묶어 읽는 docent 형식이 가장 잘 맞습니다.
참고문헌
- Jaeger, L. B., et al. “Lipopolysaccharide Alters the Blood-Brain Barrier Transport of Amyloid Beta Protein.” Brain, Behavior, and Immunity, 2009. https://pubmed.ncbi.nlm.nih.gov/19486646/
- Lee, J. W., et al. “Neuro-Inflammation Induced by Lipopolysaccharide Causes Cognitive Impairment Through Enhancement of Beta-Amyloid Generation.” Journal of Neuroinflammation, 2008. https://pubmed.ncbi.nlm.nih.gov/18759972/
- Erickson, M. A., et al. “Lipopolysaccharide Impairs Amyloid Beta Efflux From Brain.” Journal of Neuroinflammation, 2012. https://pubmed.ncbi.nlm.nih.gov/22747709/
- Zhan, X., et al. “Gram-Negative Bacterial Molecules Associate With Alzheimer Disease Pathology.” Neurology, 2016. https://pubmed.ncbi.nlm.nih.gov/27784770/
- Lukiw, W. J. “Bacteroides fragilis Lipopolysaccharide and Inflammatory Signaling in Alzheimer’s Disease.” Frontiers in Aging Neuroscience, 2018. https://pubmed.ncbi.nlm.nih.gov/29520228/
- André, C., et al. “Lipopolysaccharide-Binding Protein, Soluble CD14, and Long-Term Risk of Alzheimer Disease.” Neurology, 2019. https://pubmed.ncbi.nlm.nih.gov/31450497/
- Hunter, R. L., et al. “Lipopolysaccharide-Induced Endotoxemia Causes Alzheimer-Like Changes in Rat Brain.” Experimental Neurology, 2018. https://pubmed.ncbi.nlm.nih.gov/29755842/
- Brown, G. C., and M. T. Heneka. “The Endotoxin Hypothesis of Alzheimer’s Disease.” Molecular Neurodegeneration, 2024. https://pmc.ncbi.nlm.nih.gov/articles/PMC10983749/
- Andreadou, E. G., Katsipis, G., Tsolaki, M., and Pantazaki, A. A. “Involvement and Relationship of Bacterial Lipopolysaccharides and Cyclooxygenases Levels in Alzheimer’s Disease and Mild Cognitive Impairment Patients.” Journal of Neuroimmunology, 2021. https://doi.org/10.1016/j.jneuroim.2021.577561